进阶教程¶
主要讲述RDKit中不常见但很重要的功能以及一些方法的参数细节。 根据笔者或者读者在实际工作中的需求通过RDKit进行编写的实践代码,进一步提高工作和研发效率。
绘图¶
绘制不带颜色的分子图片¶
opts.elemDict这个参数可以控制不同元素的颜色,
def defaultDrawOptions():
'''This function returns an RDKit drawing options object with
default drawing options.'''
opts = Draw.DrawingOptions()
# opts.elemDict = defaultdict(lambda: (0,0,0)) # all atoms are black
opts.noCarbonSymbols = True
opts.selectColor = (1, 0, 0)
opts.wedgeBonds = True
opts.elemDict = defaultdict(lambda: (0, 0, 0))
opts.dotsPerAngstrom = 20
opts.bondLineWidth = 1.5
atomLabelFontFace = 'arial'
return opts
options=defaultDrawOptions()
opts.elemDict = defaultdict(lambda: (0, 0, 0))
设置成(0,0,0)代表每个元素的颜色都为黑色。
RDkit 中默认元素的颜色:
import rdkit.Chem.Draw as Draw
opts2 = Draw.DrawingOptions()
print(opts2.elemDict)
输出:
{1: (0.55, 0.55, 0.55), # H
7: (0, 0, 1), # N
8: (1, 0, 0), # O
9: (0.2, 0.8, 0.8), # F
15: (1, 0.5, 0), # P
16: (0.8, 0.8, 0), # S
17: (0, 0.8, 0), # Cl
35: (0.5, 0.3, 0.1), # Br
53: (0.63, 0.12, 0.94), # I
0: (0.5, 0.5, 0.5)} # ?
示例代码
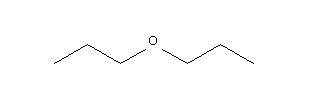
import rdkit.Chem.Draw as Draw
mol= Chem.MolFromSmiles('CCCOCCC')
opts1 = Draw.DrawingOptions()
opts1.elemDict = defaultdict(lambda: (0, 0, 0))
img1 = Draw.MolToImage(mol, options=opts1)
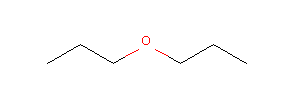
opts2 = Draw.DrawingOptions()
img2_color = Draw.MolToImage(mol, options=opts2)
display(img1)
display(img2_color)
输出:
高亮原子和键的颜色 d2d.DrawMolecule¶
我看到子结构匹配的图片时候,感觉非常漂亮,于是看了一下源码知道其是如何绘制高亮图片的。 主要是借助了**d2d.DrawMolecule**函数,里面参数有高亮的atom id列表,bond id列表, 以及自定义atom背景颜色和bond背景颜色的字典。我对其简单封装成了clourMol函数。
示例代码如下:
from rdkit import Chem
from rdkit.Chem.Draw import rdMolDraw2D
from io import BytesIO
import numpy as np
from PIL import Image, ImageOps
def _drawerToImage(d2d):
try:
import Image
except ImportError:
from PIL import Image
sio = BytesIO(d2d.GetDrawingText())
return Image.open(sio)
def clourMol(mol,highlightAtoms_p=None,highlightAtomColors_p=None,highlightBonds_p=None,highlightBondColors_p=None,sz=[400,400]):
'''
'''
d2d = rdMolDraw2D.MolDraw2DCairo(sz[0], sz[1])
op = d2d.drawOptions()
op.dotsPerAngstrom = 20
op.useBWAtomPalette()
mc = rdMolDraw2D.PrepareMolForDrawing(mol)
d2d.DrawMolecule(mc, legend='', highlightAtoms=highlightAtoms_p,highlightAtomColors=highlightAtomColors_p, highlightBonds= highlightBonds_p,highlightBondColors=highlightBondColors_p)
d2d.FinishDrawing()
product_img=_drawerToImage(d2d)
return product_img
def StripAlphaFromImage(img):
'''This function takes an RGBA PIL image and returns an RGB image'''
if len(img.split()) == 3:
return img
return Image.merge('RGB', img.split()[:3])
def TrimImgByWhite(img, padding=10):
'''This function takes a PIL image, img, and crops it to the minimum rectangle
based on its whiteness/transparency. 5 pixel padding used automatically.'''
# Convert to array
as_array = np.array(img) # N x N x (r,g,b,a)
# Set previously-transparent pixels to white
if as_array.shape[2] == 4:
as_array[as_array[:, :, 3] == 0] = [255, 255, 255, 0]
as_array = as_array[:, :, :3]
# Content defined as non-white and non-transparent pixel
has_content = np.sum(as_array, axis=2, dtype=np.uint32) != 255 * 3
xs, ys = np.nonzero(has_content)
# Crop down
margin = 5
x_range = max([min(xs) - margin, 0]), min([max(xs) + margin, as_array.shape[0]])
y_range = max([min(ys) - margin, 0]), min([max(ys) + margin, as_array.shape[1]])
as_array_cropped = as_array[
x_range[0]:x_range[1], y_range[0]:y_range[1], 0:3]
img = Image.fromarray(as_array_cropped, mode='RGB')
return ImageOps.expand(img, border=padding, fill=(255, 255, 255))
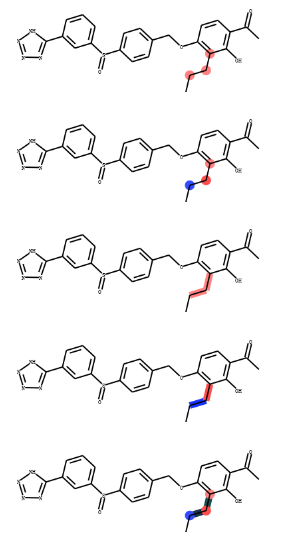
smi = 'CCCc1c(OCc2ccc(S(=O)c3cccc(-c4nnn[nH]4)c3)cc2)ccc(C(C)=O)c1O'
mol = Chem.MolFromSmiles(smi)
#hight atoms
img1 = clourMol(mol,highlightAtoms_p=[1,2,3])
img1 =StripAlphaFromImage(img1)
img1 = TrimImgByWhite(img1)
display(img1)
# custom atom color
img2=clourMol(mol,highlightAtoms_p=[1,2,3],highlightAtomColors_p={1:(0.2,0.3,1),2:(1,0.3,0.3)})
img2 =StripAlphaFromImage(img2)
img2 = TrimImgByWhite(img2)
display(img2)
# hight bond
img3 = clourMol(mol,highlightBonds_p=[1,2])
img3 =StripAlphaFromImage(img3)
img3 = TrimImgByWhite(img3)
display(img3)
# custom bond color
img4 = clourMol(mol,highlightBonds_p=[1,2],highlightBondColors_p={1:(0.1,0.2,1),2:(1,0.3,0.3)})
img4 = TrimImgByWhite(img4)
display(img4)
#all
img5 = clourMol(mol,highlightAtoms_p=[1,2,3],highlightAtomColors_p={1:(0.2,0.3,1),2:(1,0.3,0.3)},highlightBonds_p=[1,2],highlightBondColors_p={1:(0.1,0.2,0.3),2:(0.1,0.3,0.3)})
img5 = TrimImgByWhite(img5)
display(img5)
输出:
片段拼接¶
任意数目片段的拼接(最终版本)¶
最新版本下载地址:
202509月更新,最新版本是v4 版本, 增加进度条功能。
pyinstaller --onefile AutoFrag2mol_v4.py
通过网盘分享的文件:AutoFrag2mol_v4.zip
通过网盘分享的文件:AutoFrag2mol_v4.zip
链接: https://pan.baidu.com/s/18FjcFFRRwuRMwz25ZQ9mVw?pwd=gswk 提取码: gswk
--来自百度网盘超级会员v7的分享
202508月更新,最新版本是v3 版本。,
pyinstaller --onefile AutoFrag2mol_v3.py
通过网盘分享的文件:AutoFrag2mol_v3.zip
链接: https://pan.baidu.com/s/19zmCLE_GmacbxWmMZIdBbQ?pwd=wca9 提取码: wca9
--来自百度网盘超级会员v7的分享
药物化学家需要对多个片段进行系统排列组合研究,比如研究的分子由3-6个片段组成。 假设A有10种片段、B有8种片段、C有10种片段,则一共有800种组合,化学家手画需要大量的时间,利用rdkit编写脚本可以节约 化学家大量的时间。
为了方便化学家的使用,我把脚本打包成了EXE程序 AutoFrag2mol_v1.exe 。
使用说明如下:
- 创建一个工作文件夹,比如”202408”;
1. 在工作文件夹下创建文本文件, 比如有1个母核和6个R基团,则创建7个文件, 分别命名Core.txt;R1.txt;R2.txt;R3.txt;R4.txt;R5.txt;R6.txt。 这里我以6取代的苯环为例,进行演示。 在ChemDraw中绘制母核,然后复制成SMILES格式;粘贴到Core.txt文件
Core.txt
[R1]C1=C(C([R3])=C(C([R5])=C1[R6])[R4])[R2]
如果有多个母核就粘贴多个母核,一行一条SMILES;
同样的方法,创建R1到R6的txt文件,这里以R1为例。
R1.txt
[R1]C1=CC=CC=C1C#N
[R1]C1=CC=C(C#N)C=C1
- 将程序“AutoFrag2mol_v1.exe”或者脚本“AutoFrag2mol_v1.py” 拷贝到工作文件夹,双击运行程序或者运行脚本;
- 如果产生result.sdf 和 result.smi且分子数目是对的,则说明是对的。否则可以联系作者。
AutoFrag2mol_v1.exe 程序和脚本以及示例文件获取 :
通过网盘分享的文件:AutoFrag2mol_v1.zip
链接: https://pan.baidu.com/s/1qbAVcr5xmOX__mAVGXxOPg?pwd=krdz 提取码: krdz
--来自百度网盘超级会员v6的分享
AutoFrag2mol_v1.py 脚本
# author: Zhaoqiang Chen
# contact: 18321885908
from glob import glob
import itertools
import re
from rdkit import Chem
from rich import print
def txt2rdkitsmi(txtf):
'''
'''
fh = open(txtf)
smis = []
for line in fh:
if line:
chemsmi = line.strip()
# Replace R groups with "*:"
smi = re.sub(r'R', '*:', chemsmi)
smis.append(smi)
return smis
def frags2mols(allFrags_smi):
'''
'''
# 使用 itertools.product 生成所有排列组合
combinations = list(itertools.product(*allFrags_smi))
# 输出结果
results=[]
for combination in combinations:
# print(combination)
smi = '.'.join(combination)
mol = Chem.MolFromSmiles(smi)
mol_com = Chem.molzip(mol)
newsmi = Chem.MolToSmiles(mol_com)
# print(newsmi)
results.append(newsmi)
return results
def AutoFrag2Mol():
'''
author: Zhaoqiang Chen
contact: 18321885908
'''
print("Welcome to use the Programe AutoFrag2mol")
print("Manual: http://rdkit.chenzhaoqiang.com/mediaManual.html#id5")
print("If you have problems,please contact Me")
msg = '''
author: Zhaoqiang Chen
contact: 18321885908
'''
print("Contact:",msg)
txtfiles = glob("*txt")
#txt2rdkit_smi
allFrags_smi=[]
for txtf in txtfiles:
rdkitsmiles = txt2rdkitsmi(txtf)
allFrags_smi.append(rdkitsmiles)
smis = frags2mols(allFrags_smi)
results =smis
print("total generated %s smiles"%len(results))
results_uniq=list(set(results))
print("total generated uniq %s smiles"%len(results_uniq))
fw = open('result.txt','w')
w = Chem.SDWriter("result.sdf")
for smi in results_uniq:
fw.write(smi+"\n")
mol = Chem.MolFromSmiles(smi)
w.write(mol)
w.close()
fw.close()
print("Results are save to the %s and %s"%("result.txt","result.sdf"))
print("If it help you, you could donation to me in the website")
print("http://rdkit.chenzhaoqiang.com/donation.html")
if __name__=='__main__':
'''
'''
AutoFrag2Mol()
2025.05 增加了v2 版本,1. 查看哪些分子重复;2. 检查有问题的字符串;3. 自动删除SMILES 字符串中文本的空行。
通过网盘分享的文件:AutoFrag2mol_v2.exe
链接: https://pan.baidu.com/s/19oUfdJqbGS-ko3NvxR5vaQ?pwd=r7r2 提取码: r7r2
--来自百度网盘超级会员v6的分享
# author: Zhaoqiang Chen
# contact: 18321885908
from glob import glob
import itertools
import re
from rdkit import Chem
from rich import print
from collections import Counter
def txt2rdkitsmi(txtf):
'''
'''
fh = open(txtf)
smis = []
for line in fh:
line = line.strip()
if line:
chemsmi = line.strip()
# Replace R groups with "*:"
smi = re.sub(r'R', '*:', chemsmi)
smis.append(smi)
return smis
def frags2mols(allFrags_smi):
'''
'''
# 使用 itertools.product 生成所有排列组合
combinations = list(itertools.product(*allFrags_smi))
# 输出结果
results=[]
for combination in combinations:
smi = '.'.join(combination)
mol = Chem.MolFromSmiles(smi)
try:
mol_com = Chem.molzip(mol)
except:
print(combination)
print(newsmi)
print("smi:",smi)
raise("stop ERROR")
newsmi = Chem.MolToSmiles(mol_com)
results.append(newsmi)
return results
def AutoFrag2Mol():
'''
author: Zhaoqiang Chen
contact: 18321885908
'''
print("Welcome to use the Programe AutoFrag2mol")
print("Manual: http://rdkit.chenzhaoqiang.com/mediaManual.html#id5")
print("If you have problems,please contact Me")
msg = '''
author: Zhaoqiang Chen
contact: 18321885908
'''
print("Contact:",msg)
txtfiles = glob("*txt")
#txt2rdkit_smi
allFrags_smi=[]
for txtf in txtfiles:
rdkitsmiles = txt2rdkitsmi(txtf)
allFrags_smi.append(rdkitsmiles)
smis = frags2mols(allFrags_smi)
results =smis
count = Counter(results)
# 只打印出现次数 >1 的元素
resu_more = {k: v for k, v in count.items() if v != 1}
print("出现超过1次的结果:")
print(resu_more) # 输出:{2: 2, 3: 3}
print("total generated %s smiles"%len(results))
results_uniq=list(set(results))
print("total generated uniq %s smiles"%len(results_uniq))
fw = open('result.txt','w')
w = Chem.SDWriter("result.sdf")
for smi in results_uniq:
fw.write(smi+"\n")
mol = Chem.MolFromSmiles(smi)
w.write(mol)
w.close()
fw.close()
print("Results are save to the %s and %s"%("result.txt","result.sdf"))
print("If it help you, you could donation to me in the website")
print("http://rdkit.chenzhaoqiang.com/donation.html")
# Pause the console
input("Press Enter to exit...")
if __name__=='__main__':
'''
'''
AutoFrag2Mol()
2025.08 增加了v3 版本,1. 增加了母核中包含2个相同基团的场景。
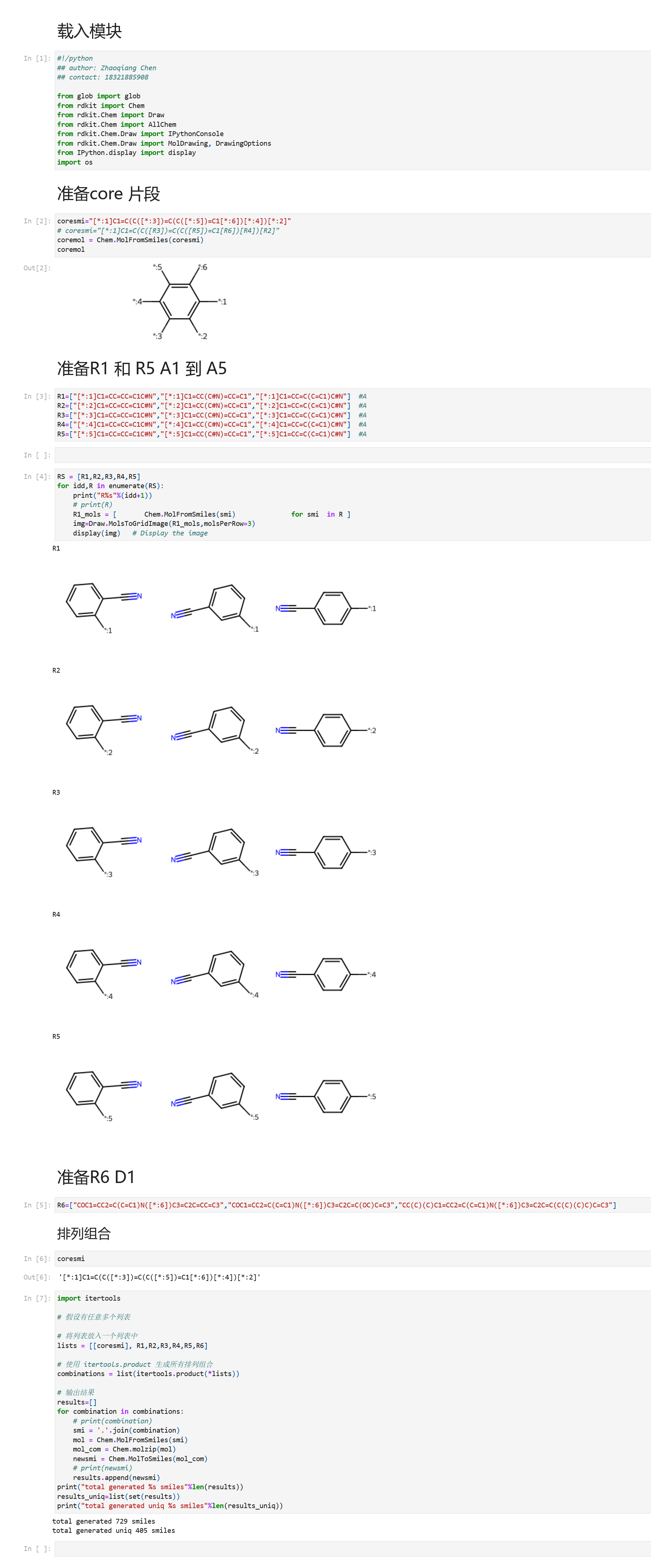
示例母核如下:
使用方法相同,准备core.txt, R1.txt, R2.txt 。
pyinstaller --onefile AutoFrag2mol_v3.py
通过网盘分享的文件:AutoFrag2mol_v3.zip
链接: https://pan.baidu.com/s/19zmCLE_GmacbxWmMZIdBbQ?pwd=wca9 提取码: wca9
--来自百度网盘超级会员v7的分享
# author: Zhaoqiang Chen
# contact: 18321885908
from glob import glob
import itertools
import re
from rdkit import Chem
from rich import print
from collections import Counter
def txt2rdkitsmi(txtf):
'''
'''
fh = open(txtf)
smis = []
for line in fh:
line = line.strip()
if line:
chemsmi = line.strip()
# Replace R groups with "*:"
smi = re.sub(r'R', '*:', chemsmi)
smis.append(smi)
return smis
def selectcore(combination):
count_num=0
for smi in combination:
# 匹配所有 [*:n] 格式的标记原子
matches = re.findall(r'\[\*:\d\]', smi)
# 统计总数
count_num = len(matches)
if count_num > 1:
return smi
if count_num == 1:
return combination[0]
raise "ERROR not found core smi"
def frags2mols(allFrags_smi):
'''
'''
# 使用 itertools.product 生成所有排列组合
combinations = list(itertools.product(*allFrags_smi))
# 输出结果
results=[]
for combination in combinations:
core = selectcore(combination)
matches = re.findall(r'\[\*:\d\]',core )
core_num = len(matches)
if core_num+1 == len(combination):
smi = '.'.join(combination)
mol = Chem.MolFromSmiles(smi)
try:
mol_com = Chem.molzip(mol)
except:
print(combination)
print(newsmi)
print("smi:",smi)
raise("stop ERROR")
elif core_num+1 > len(combination):
# print("core",core)
# print("combination",combination)
combination_copy = list(combination)
combination_copy.remove(core)
rgroups = combination_copy
# print("rgroups",rgroups)
# core_mol = Chem.MolFromSmiles(core)
# bignum = matches = re.findall(r'\[\*:\d\]', core )
numbers = [int(match.group(1)) for match in re.finditer(r'\[\*:(\d+)\]', core)]
bignum = max(numbers)
newrgoups=[]
for rgroup in rgroups:
# print("rgroups",rgroups)
# print("rgroup",rgroup)
matches = re.findall(r'\[\*:\d\]', rgroup)
num = len(matches)
if num == 1:
rgroup_num = matches[0]
count = core.count(rgroup_num)
newrgoups.append(rgroup)
if count > 1:
matches = list(re.finditer(rgroup_num, core))
for i in range(count-1):
new_num = 1 + bignum
bignum = new_num
new_str = f'[*:{new_num}]'
# core = core[:match.start()] + new_str + core[match.end():]
# print("core before",core )
# print("dddddd",rgroup_num, '[*:%d]'%bignum,"!!!!!!!!")
pattern = re.escape(rgroup_num)
core = re.sub(pattern , '[*:%d]'%bignum, core,count=1)
# print("core after",core )
# raise "stop0"
# print("rgroup_num",rgroup_num) # [*:1]
pattern = re.escape(rgroup_num)
new_smi = re.sub(pattern, '[*:%d]'%bignum, rgroup,count=1)
temp_group = new_smi
# print("new_smi",new_smi)
# print("core",core)
# raise "stop"
newrgoups.append(temp_group)
# print("更新后的 SMILES:", core)
else:
print("[*:1] 出现次数 ≤ 2,无需更新")
pass
else:
raise "ERROR: Rgroup have more than one group"
combination=[core]+newrgoups
smi = '.'.join(combination)
# print("result smi",smi)
# raise "stop"
mol = Chem.MolFromSmiles(smi)
mol_com = Chem.molzip(mol)
newsmi = Chem.MolToSmiles(mol_com)
results.append(newsmi)
return results
def AutoFrag2Mol():
'''
author: Zhaoqiang Chen
contact: 18321885908
'''
print("Welcome to use the Programe AutoFrag2mol")
print("Manual: http://rdkit.chenzhaoqiang.com/mediaManual.html#id5")
print("If you have problems,please contact Me")
msg = '''
author: Zhaoqiang Chen
contact: 18321885908
'''
print("Contact:",msg)
txtfiles = glob("*txt")
#txt2rdkit_smi
allFrags_smi=[]
for txtf in txtfiles:
rdkitsmiles = txt2rdkitsmi(txtf)
allFrags_smi.append(rdkitsmiles)
smis = frags2mols(allFrags_smi)
results =smis
count = Counter(results)
# 只打印出现次数 >1 的元素
resu_more = {k: v for k, v in count.items() if v != 1}
print("出现超过1次的结果:")
print(resu_more) # 输出:{2: 2, 3: 3}
print("total generated %s smiles"%len(results))
results_uniq=list(set(results))
print("total generated uniq %s smiles"%len(results_uniq))
fw = open('result.txt','w')
w = Chem.SDWriter("result.sdf")
for smi in results_uniq:
fw.write(smi+"\n")
mol = Chem.MolFromSmiles(smi)
w.write(mol)
w.close()
fw.close()
print("Results are save to the %s and %s"%("result.txt","result.sdf"))
print("If it help you, you could donation to me in the website")
print("http://rdkit.chenzhaoqiang.com/donation.html")
# Pause the console
input("Press Enter to exit...")
if __name__=='__main__':
'''
'''
AutoFrag2Mol()
3片段拼接(废弃)¶
从实用的角度只需要看 任意数目片段的拼接(最终版本)
药物化学家需要对多个片段进行系统排列组合研究,比如研究的分子由3个片段(A片段,B片段,C片段)组成。 假设A有10种片段、B有8种片段、C有10种片段,则一共有800种组合,化学家手画需要大量的时间,利用rdkit编写脚本可以节约 化学家大量的时间。
该脚本也适用于Protac的拼接功能。
编写代码combine3_v1.py, 代码如下:
#!/python
## author: Zhaoqiang Chen
## contact: 18321885908
from glob import glob
from rdkit import Chem
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem.Draw import MolDrawing, DrawingOptions
def combine3frag(Amol,Bmol,Cmol):
ABmolsmi=combine2frag(Amol,'Fr',Bmol,'Cs')
print('ABSMI',ABmolsmi)
ABmol= Chem.MolFromSmiles(ABmolsmi)
ABCmolsmi=combine2frag(ABmol,'Rb',Cmol,'Fr')
print("ABCsmi","ABCmolsmi")
ABCmol=Chem.MolFromSmiles(ABCmolsmi)
smi=Chem.MolToSmiles(ABCmol)
return smi
def combine2frag(Amol,Fr,Bmol,Cs):
combo = Chem.CombineMols(Amol,Bmol)
Fr_NEI_ID=get_neiid_bysymbol(combo,Fr)
Cs_NEI_ID=get_neiid_bysymbol(combo,Cs)
edcombo = Chem.EditableMol(combo)
edcombo.AddBond(Fr_NEI_ID,Cs_NEI_ID,order=Chem.rdchem.BondType.SINGLE)
Fr_ID=get_id_bysymbol(combo,Fr)
edcombo.RemoveAtom(Fr_ID)
back = edcombo.GetMol()
Cs_ID=get_id_bysymbol(back,Cs)
edcombo=Chem.EditableMol(back)
edcombo.RemoveAtom(Cs_ID)
back = edcombo.GetMol()
smi= Chem.MolToSmiles(back)
return smi
def get_neiid_bysymbol(combo,symbol):
for at in combo.GetAtoms():
if at.GetSymbol()==symbol:
at_nei=at.GetNeighbors()[0]
return at_nei.GetIdx()
def get_id_bysymbol(combo,symbol):
for at in combo.GetAtoms():
if at.GetSymbol()==symbol:
return at.GetIdx()
Asdfs=glob('./A/*')
Bsdfs=glob('./B/*')
Csdfs=glob('./C/*')
Asdfs=glob('./A/*')
Bsdfs=glob('./B/*')
Csdfs=glob('./C/*')
Amols=[ Chem.SDMolSupplier(f)[0] for f in Asdfs]
Bmols=[ Chem.SDMolSupplier(f)[0] for f in Bsdfs]
Cmols=[ Chem.SDMolSupplier(f)[0] for f in Csdfs]
resultsmi=[]
for Amol in Amols[0:]:
for Bmol in Bmols[0:]:
for Cmol in Cmols[0:]:
smi=combine3frag(Amol,Bmol,Cmol)
print(smi)
resultsmi.append(smi)
w = Chem.SDWriter("result.sdf")
for smi in resultsmi:
mol = Chem.MolFromSmiles(smi)
w.write(mol)
w.close()
为了方便化学家的使用,我把脚本打包成了EXE程序 combine3_v1.exe 。
使用说明如下:
- 创建一个工作文件夹,比如”20220430”;
- 在工作文件夹下分别创建3个文件夹, 并命名为”A”,”B”,”C”;
- 用chemdraw 编辑A片段,连接的地方用’Fr’原子表示,保存为sdf文件,如A1.sdf,A2.sdf,A3.sdf等
- 用chemdraw 编辑B片段,连接的地方用’Cs’ ‘Rb’原子表示,保存为sdf文件,Cs接头和A片段的Fr接头项链,如B1.sdf,B2.sdf,B3.sdf等
- 用chemdraw 编辑C片段,连接的地方用’Fr’原子表示,Fr接头和B片段的Rb接头相连,保存为sdf文件,如C1.sdf,C2.sdf,B3.sdf等
- 将程序“combine3_v1.exe”或者脚本“combine3_v1.py” 拷贝到工作文件夹,双击运行程序或者运行脚本;
- 如果产生result.sdf且分子数目是对的,则说明是对的。否则可以联系作者。
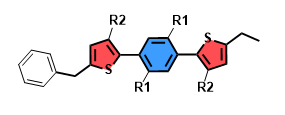
示例A片段如下:
A1: [Fr]c1ccccc1
A2: Cc1cccc([Fr])c1
A3: CCc1cccc([Fr])c1
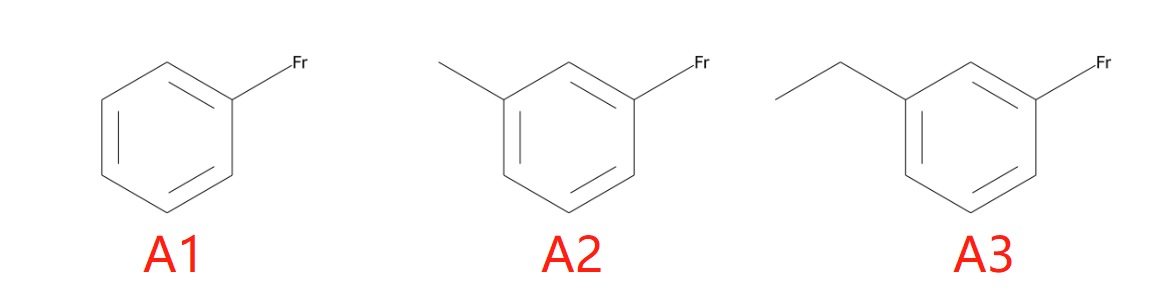
示例B片段如下:
B1: [Rb]CC1CCC(C[Cs])CC1
B2: O=C([Rb])C1CCC(C[Cs])CC1
B3: O=C([Rb])C1CCC(C2([Cs])CC2)CC1
示例C片段如下:
C1: [Fr]C1CC1
C2: CC1CC1[Fr]
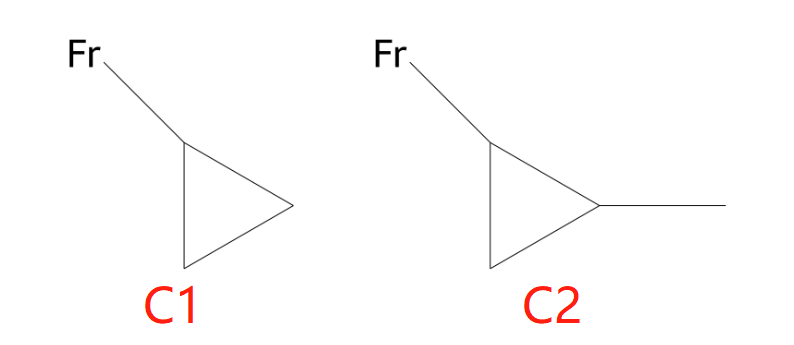
拼接后分子如下:
百度网盘下载连接如下:
链接:https://pan.baidu.com/s/1Mdi8E7ElAVjvnf242M02Ew?pwd=xa51
提取码:xa51
--来自百度网盘超级会员V4的分享
2片段(多片段)拼接¶
不管是3片段还是4片段的拼接本质上可以看成是2片段的拼接, 比如A-B-C-D的拼接,我们可以先让A和B拼接得到AB片段, 再让AB和C拼接得到ABC片段,最后让ABC和D 拼接得到ABCD。
为了增加代码的灵活性,这里引入了配置文件config.ini文件。 第一行中填入A片段的接头符号和B片段的接头符号,以空格隔开。
配置文件(config.ini)如下:
Fr Cs
代码如下:
#!/python
## author: Zhaoqiang Chen
## contact: 18321885908
from glob import glob
from rdkit import Chem
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem.Draw import MolDrawing, DrawingOptions
import os
# if os.path.exists('AB'):
# raise("please delete or rename AB dir")
# else:
# os.mkdir("AB")
def combine2frag(Amol,Fr,Bmol,Cs):
combo = Chem.CombineMols(Amol,Bmol)
Fr_NEI_ID=get_neiid_bysymbol(combo,Fr)
Cs_NEI_ID=get_neiid_bysymbol(combo,Cs)
edcombo = Chem.EditableMol(combo)
edcombo.AddBond(Fr_NEI_ID,Cs_NEI_ID,order=Chem.rdchem.BondType.SINGLE)
Fr_ID=get_id_bysymbol(combo,Fr)
edcombo.RemoveAtom(Fr_ID)
back = edcombo.GetMol()
Cs_ID=get_id_bysymbol(back,Cs)
edcombo=Chem.EditableMol(back)
edcombo.RemoveAtom(Cs_ID)
back = edcombo.GetMol()
smi= Chem.MolToSmiles(back)
return smi
def get_neiid_bysymbol(combo,symbol):
for at in combo.GetAtoms():
if at.GetSymbol()==symbol:
at_nei=at.GetNeighbors()[0]
return at_nei.GetIdx()
def get_id_bysymbol(combo,symbol):
for at in combo.GetAtoms():
if at.GetSymbol()==symbol:
return at.GetIdx()
Asdfs=glob('./A/*')
Bsdfs=glob('./B/*')
Asdfs=glob('./A/*')
Bsdfs=glob('./B/*')
Amols=[ Chem.SDMolSupplier(f)[0] for f in Asdfs]
Bmols=[ Chem.SDMolSupplier(f)[0] for f in Bsdfs]
resultsmi=[]
resultmols=[]
Fr='Fr'
Cs='Cs'
if os.path.exists('config.ini'):
fh=open('config.ini')
line=fh.readlines()[0].strip()
Fr,Cs =line.split()
asmi = Chem.MolToSmiles(Amols[0])
bsmi = Chem.MolToSmiles(Bmols[0])
if Fr in asmi and Cs in bsmi:
pass
else:
raise("error,please fill joint symbol into the file config.ini")
for Amol in Amols[0:]:
for Bmol in Bmols[0:]:
smi=combine2frag(Amol,Fr,Bmol,Cs)
print(smi)
resultsmi.append(smi)
w = Chem.SDWriter("result.sdf")
for smi in resultsmi:
mol = Chem.MolFromSmiles(smi)
w.write(mol)
w.close()
使用说明:
- 将程序“combine2_v1.exe”或者脚本“combine2_v1.py” 拷贝到工作文件夹,双击运行程序或者运行脚本;
- 如果产生result.sdf且分子数目是对的,则说明是对的。否则可以联系作者。
百度网盘下载连接如下
链接:https://pan.baidu.com/s/17c8Hg2SJy9myuLeJwef7AQ?pwd=58sq
提取码:58sq
DeepFrag2Mol¶
DeepFrag2Mol:把DeepFrag程序产生的分子组装成完整的分子。
注解
注意事项: 1. 母核骨架SMILES中的接头原子*,必须放在中括号中。
方法1: 源码安装
git clone https://github.com/autodataming/DeepFrag2Mol.git
cd conda env create -f deepfrag2mol.yaml
conda activate deepfrag2mol
方法2: 二进制安装
链接:https://pan.baidu.com/s/10zFsY2mWBxXzQ0Wj9aZk2g?pwd=aj9k 提取码:aj9k
用法:
python deepFrag2mol_CMDgui.py
(base) D:\DeepFrag2mol_v2024>python deepFrag2mol_CMDgui.py
DeepFrag2Mol: Converting the molecular fragments generated by DeepFrag into complete molecules.
Usage: deepFrag2mol_CMDgui.py [OPTIONS]
Options:
--scaffold TEXT the SMILES of the molecular scaffold
--fragfile TEXT the file of the molecular fragments generated by DeepFrag
--help Show this message and exit.
input scaffold SMILES: [[*]C1=CC=CC=C1]:
input fragment file path and name [deepfrag-scores.csv]:
89%|███████████████████████████████████████████████████████████████████▋ | 4953/5564 [00:00<00:00, 7662.98it/s][13:19:03] Explicit valence for atom # 1 B, 5, is greater than permitted
error: [Fr]C1=CC=CC=C1 [Cs][BH4]
100%|████████████████████████████████████████████████████████████████████████████| 5564/5564 [00:00<00:00, 7252.01it/s]
Congratulations,You have converted 5564 total fragments to 5563 molecules, The result is saved in file deepfrag-scores_mol.smi
github地址如下:
https://github.com/autodataming/DeepFrag2Mol
百度网盘链接如下:
链接:https://pan.baidu.com/s/10zFsY2mWBxXzQ0Wj9aZk2g?pwd=aj9k 提取码:aj9k
PMI分析¶
分子的空间分布分析:
文献中的示例图片:
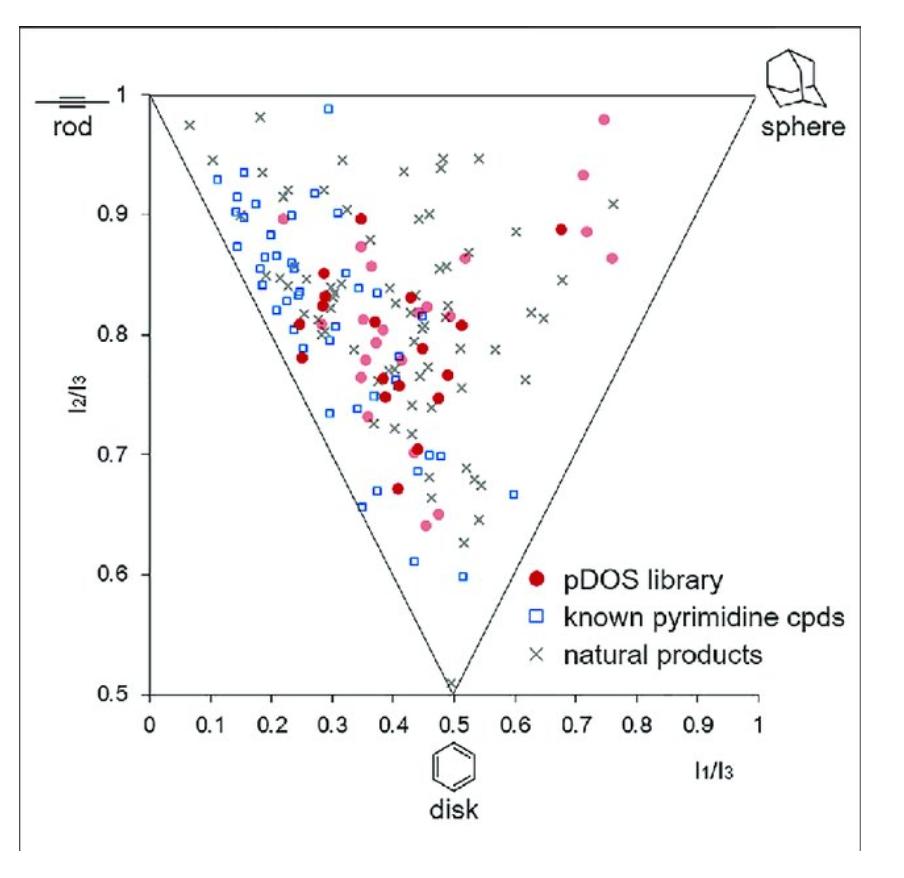
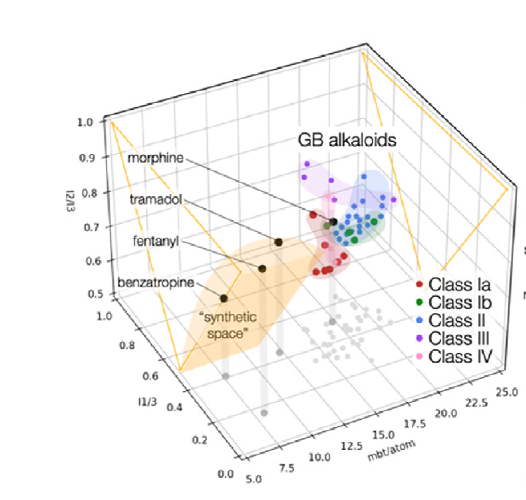
2D 图形的横坐标是Disklikeness=I1/I3;纵坐标是Rodlikeness=I2/I3;
苯(0.5,0.5);乙炔(0,1);金刚烷(1,1)
disk=x=rdkit.Chem.Descriptors3D.NPR1(*x, **y) : Normalized principal moments ratio 1 (=I1/I3)
y=rod=rdkit.Chem.Descriptors3D.NPR2(*x, **y) : Normalized principal moments ratio 2 (=I2/I3)
示例代码
import rdkit
import rdkit
from rdkit import Chem
from rdkit.Chem import AllChem
CCsmi="C#C"
bensmi="C1=CC=CC=C1"
Adamantanesmi="C1C2CC3CC1CC(C2)C3"
smi=CCsmi
mol=Chem.MolFromSmiles(smi)
m2=Chem.AddHs(mol)
AllChem.EmbedMolecule(m2)
# maxIters默认是优化200次,有时不会收敛,建议设置成2000
opt_state=AllChem.MMFFOptimizeMolecule(m2,maxIters=2000)
x=rdkit.Chem.rdMolDescriptors.CalcNPR1(m2)
y=rdkit.Chem.rdMolDescriptors.CalcNPR2(m2)
print("CC",x,y)
smi=bensmi
mol=Chem.MolFromSmiles(smi)
m2=Chem.AddHs(mol)
AllChem.EmbedMolecule(m2)
# maxIters默认是优化200次,有时不会收敛,建议设置成2000
opt_state=AllChem.MMFFOptimizeMolecule(m2,maxIters=2000)
# print(opt_state)
m2
x=rdkit.Chem.rdMolDescriptors.CalcNPR1(m2)
y=rdkit.Chem.rdMolDescriptors.CalcNPR2(m2)
print("benzene",x,y)
smi=Adamantanesmi
mol=Chem.MolFromSmiles(smi)
m2=Chem.AddHs(mol)
AllChem.EmbedMolecule(m2)
# maxIters默认是优化200次,有时不会收敛,建议设置成2000
opt_state=AllChem.MMFFOptimizeMolecule(m2,maxIters=2000)
m2
x=rdkit.Chem.rdMolDescriptors.CalcNPR1(m2)
y=rdkit.Chem.rdMolDescriptors.CalcNPR2(m2)
print("adamantane",x,y)
输出:
CC 7.614072578672083e-14 0.9999999999999301
benzene 0.4999999843169691 0.5000000156831409
adamantane 0.9999994596825732 0.9999998986910213
molecular complexity的定义不同人有不同的定义,rdkit中自带是Bertz的定义。
rdkit.Chem.GraphDescriptors.BertzCT(mol, cutoff=100, dMat=None, forceDMat=1)
A topological index meant to quantify “complexity” of molecules.
Consists of a sum of two terms, one representing the complexity of the bonding, the other representing the complexity of the distribution of heteroatoms.
From S. H. Bertz, J. Am. Chem. Soc., vol 103, 3599-3601 (1981)
文献中使用的Bottcher的定义。
注解
Bottcher molecular complexity: https://github.com/boskovicgroup/bottchercomplexity
GB18: 448.39 mcbits, 17.25 mcbits/atom; himgaline: 477.83 mcbits, 20.78 mcbits/atom
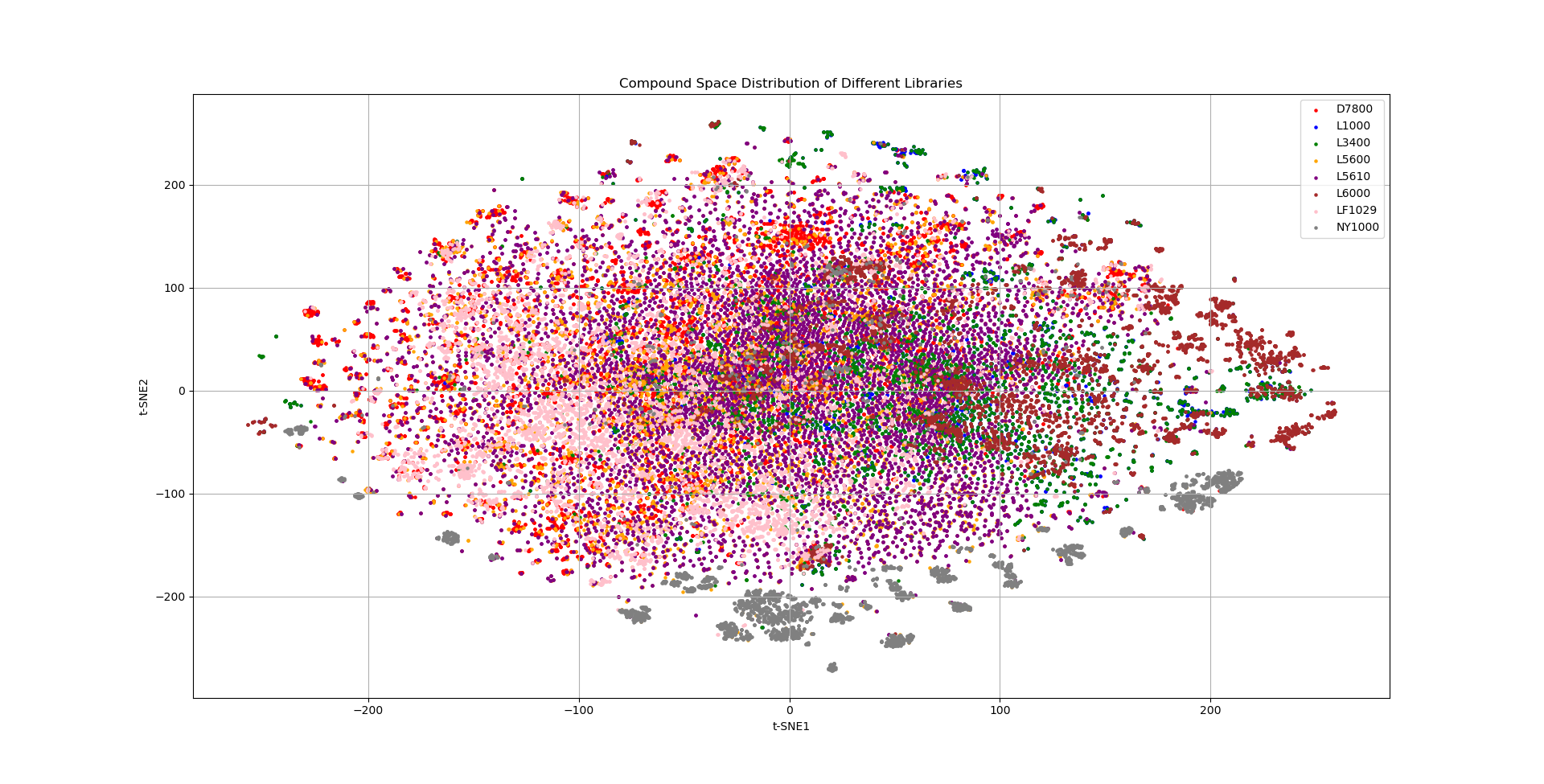
tSNE化合物库空间分析¶
有多个化合物库,想看看怎么组合使得化合物空间覆盖更广。
伪代码如下:
1. 计算所有库中分子的指纹,并转换成数组;
2. tSNE 分析;
3. 作图。
注解
1. tSNE 耗时较长,如果化合物库很大,建议先PCA降维,再做tSNE分析。 参考:https://github.com/rdkit/rdkit-tutorials/blob/master/notebooks/005_Chemical_space_analysis_and_visualization.ipynb
- 不能对每个库分别进行fit_transform分析,必须把所有库的指纹合并为一个数组,然后一起进行 t-SNE 计算。
代码如下:
from rdkit import Chem
from rdkit.Chem import Descriptors
from sklearn.manifold import TSNE
import matplotlib.pyplot as plt
import numpy as np
import os
from rdkit.Chem import rdFingerprintGenerator
from rdkit import DataStructs
from glob import glob
mfpgen = rdFingerprintGenerator.GetMorganGenerator(radius=2, fpSize=2048)
model = TSNE(n_components=2, random_state=0, perplexity=30, n_iter=5000)
def fp_from_mol(mol):
fp = mfpgen.GetFingerprint(mol)
return fp
def mols2fps(mols_gen):
fps = []
totalid = 0
errid = 0
for mol in mols_gen:
totalid += 1
try:
Chem.SanitizeMol(mol)
fp = fp_from_mol(mol)
np_array = np.zeros((1, 2048), dtype=np.int8) # Adjust size here
DataStructs.ConvertToNumpyArray(fp, np_array[0]) # Populate the first row
fps.append(np_array[0]) # Append the first row as 1D array
except Exception as e:
print("An error occurred:", str(e))
errid += 1
smi = Chem.MolToSmiles(mol) if mol else "Invalid Molecule"
print("error smi:", errid, totalid, smi)
return fps
def tSNE(sdf_files):
basenames = [i.strip('.sdf') for i in sdf_files]
fps_legend = {}
legends=[]
all_fps=[]
for sdf_file in sdf_files:
filename = sdf_file
basename, file_extension = os.path.splitext(sdf_file)
legend = basename
if file_extension == '.sdf':
mols_gen = Chem.SDMolSupplier(filename, sanitize=False)
fps_array = mols2fps(mols_gen)
fps_legend[legend] = np.array(fps_array)
legends.append(legend)
all_fps.extend(fps_array)
else:
print("%s extension format is not supported" % file_extension)
# 合并所有指纹
all_fps = np.array(all_fps)
# t-SNE 计算
tsne_results = model.fit_transform(all_fps)
plt.figure(figsize=(10, 6))
colors = ['red', 'blue', 'green', 'orange', 'purple', 'brown', 'pink', 'gray', 'cyan', 'magenta']
idd = 0
# 绘制散点图
for legend in legends:
color = colors[idd]
start_idx = sum(len(fps_legend[k]) for k in legends[:idd])
end_idx = start_idx + len(fps_legend[legend])
x = tsne_results[start_idx:end_idx, 0]
y = tsne_results[start_idx:end_idx, 1]
plt.scatter(x, y, color=color, label=legend,s=5)
idd += 1
plt.title('Compound Space Distribution of Different Libraries')
plt.xlabel('t-SNE1')
plt.ylabel('t-SNE2')
plt.legend()
plt.grid()
plt.show()
if __name__ == '__main__':
sdf_files = ['L3400.sdf', 'L5600.sdf', 'L5610.sdf']
sdf_files = glob("*sdf")
tSNE(sdf_files)
示例文件:
通过网盘分享的文件:tSNE.zip
链接: https://pan.baidu.com/s/1BCJz9ovhJfNM96d6MJHTtw?pwd=m9zx 提取码: m9zx
用法:
1. 把需要分析的sdf文件,拷贝到tSNE.py脚本所在目录;
2. (base) D:\notebookWD\molstat\tSNE>python tSNE.py
输出:
形状相似性计算¶
Dear all,
A c++ implementation and Python wrappers of the ultrafast shape recognition
(USR) descriptor (Ballester and Richards, J. Comput. Chem. (2007), 28,
1711) and the USR CREDO atom types (USRCAT) descriptor (Schreyer and
Blundell, J. Cheminf. (2012), 4, 27) are now available for the RDKit. The
code is based on the Python implementations of Jan Domanski and Adrian
Schreyer.
The descriptors can be accessed from Python via
rdkit.Chem.rdMolDescriptors.GetUSR and
rdkit.Chem.rdMolDescriptors.GetUSRCAT.
Best regards,
Sereina
应用场景1:
分子生成-》对接-》相互作用-》形状过滤-》设计分子
应用场景2
2D smiles->3D mol->minization-> 形状的相似性打分
示例文件 lincs_50_smile.txt 。
示例代码:
import rdkit
from rdkit import Chem
from rdkit.Chem.Descriptors import NumValenceElectrons
from rdkit.Chem import Descriptors
from rdkit.Chem import AllChem
from rdkit.Chem.rdMolDescriptors import GetUSRScore, GetUSRCAT
import nglview
fh=open('lincs_50_smile.txt')
smis=[]
for line in fh.readlines():
smi=line.strip()
smis.append(smi)
# print(smis)
mols3d=[]
for smi in smis:
mol=Chem.MolFromSmiles(smi)
m2=Chem.AddHs(mol)
AllChem.EmbedMolecule(m2)
# maxIters默认是优化200次,有时不会收敛,建议设置成2000
opt_state=AllChem.MMFFOptimizeMolecule(m2,maxIters=2000)
# print(opt_state)
mols3d.append(m2)
usrcats = [ GetUSRCAT( mol ) for mol in mols3d ]
for i in range( len( usrcats )):
for j in range( len( usrcats )):
score = GetUSRScore( usrcats[ i ], usrcats[ j ] )
print(i,j,"usrscroe:",score)
输出:
0 0 usrscroe: 1.0
0 1 usrscroe: 0.2136718008888286
0 2 usrscroe: 0.11508212828197005
0 3 usrscroe: 0.11843775593882168
0 4 usrscroe: 0.1447975173956143
0 5 usrscroe: 0.12813777238473245
0 6 usrscroe: 0.13726495667508753
0 7 usrscroe: 0.12131620151077707
0 8 usrscroe: 0.0900556733174051
0 9 usrscroe: 0.11663586460430561
注解
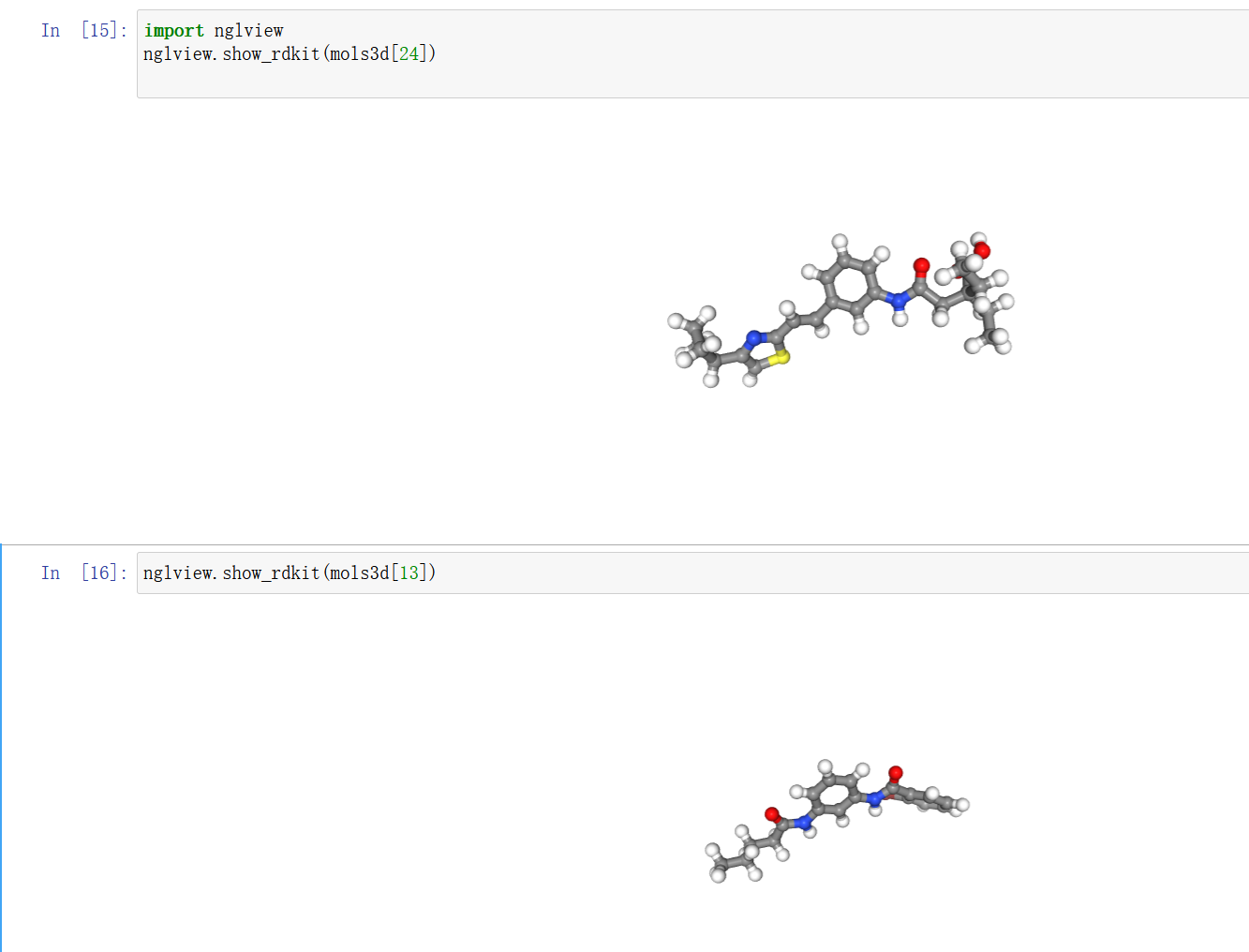
jupyter 文档中可以使用nglview 交互查看分子的3D 构象
import nglview
nglview.show_rdkit(mols3d[24])
nglview.show_rdkit(mols3d[13])
USR 描述符¶
- ultrafast shape recognition
- (USR) descriptor
https://iwatobipen.wordpress.com/2017/11/17/calculate-usrcat-with-rdkit-rdkit/
USRCAT 描述符¶
USR CREDO atom types (USRCAT) descriptor
https://iwatobipen.wordpress.com/2017/11/17/calculate-usrcat-with-rdkit-rdkit/
首尾相连环肽切割肽键获得氨基酸残基¶
首尾相连环肽切割肽键获得氨基酸残基,
示例文件:
示例代码:
import rdkit
from rdkit import Chem
from rdkit.Chem import Draw
def break_cyclic_peptide_advanced(mol):
"""更精确的环肽切割方法 - 修正版"""
# 定义酰胺键模式:环上的羰基碳连接环上的氮原子
amide_smarts = [
'[C;R](=[O])-[#7;R]', # 最准确的酰胺键模式
]
bonds_to_break = []
for pattern_smarts in amide_smarts:
pattern = Chem.MolFromSmarts(pattern_smarts)
if pattern is None:
print(f"无效的SMARTS模式: {pattern_smarts}")
continue
matches = mol.GetSubstructMatches(pattern)
print(f"模式 '{pattern_smarts}' 找到 {len(matches)} 个匹配")
for i, match in enumerate(matches):
print(f"匹配 {i+1}: 原子索引 {match}")
# 在匹配的原子中寻找C-N键
# match[0]是环上的碳原子,match[2]是环上的氮原子(因为模式是C(=O)-N)
if len(match) >= 3:
carbon_idx = match[0] # 羰基碳原子
nitrogen_idx = match[2] # 氮原子
# 获取两个原子
carbon_atom = mol.GetAtomWithIdx(carbon_idx)
nitrogen_atom = mol.GetAtomWithIdx(nitrogen_idx)
# 验证原子类型和环属性
if (carbon_atom.GetSymbol() == 'C' and nitrogen_atom.GetSymbol() == 'N' and
carbon_atom.IsInRing() and nitrogen_atom.IsInRing()):
# 找到连接这两个原子的键
bond = mol.GetBondBetweenAtoms(carbon_idx, nitrogen_idx)
if bond is not None:
bond_idx = bond.GetIdx()
bonds_to_break.append(bond_idx)
print(f" 找到酰胺键: C{carbon_idx}-N{nitrogen_idx} (键索引: {bond_idx})")
else:
print(f" 错误: 原子 {carbon_idx} 和 {nitrogen_idx} 之间没有键")
else:
print(f" 验证失败: C{carbon_idx}(环上:{carbon_atom.IsInRing()}), N{nitrogen_idx}(环上:{nitrogen_atom.IsInRing()})")
# 去重
bonds_to_break = list(set(bonds_to_break))
print(f"最终要切割的键: {bonds_to_break}")
if bonds_to_break:
print(f"将在 {len(bonds_to_break)} 个酰胺键处切割")
# 添加虚拟原子以便后续处理
fragmented_mol = Chem.FragmentOnBonds(mol, bonds_to_break,
addDummies=True,
dummyLabels=[(1, 1)] * len(bonds_to_break))
fragments = Chem.GetMolFrags(fragmented_mol, asMols=True, sanitizeFrags=True)
return fragments
print("未找到符合条件的酰胺键")
return [mol]
smi="CC[C@H](C)[C@H]([N]C([C@H](CCCC[N])[N]C([C@H](CCC[N]C([N])=[N])[N]C(C([N]C1=O)Cc2c[nH]cn2)=O)=O)=O)C([N][C@@H](Cc3ccccc3)C([N][C@@H](CC(C)C)C([N][C@@H](Cc4c5ccccc5[nH]c4)C([N][C@@H](C)C([N][C@@H](CCSC)C(N6CCC[C@H]6C([N][C@@H](C[S])C([N][C@@H](CC([N])=O)C([N][C@H](C([N]CC([N][C@@H](C[O])C([N][C@@H](CCC([N])=O)C([N][C@@H](Cc7ccc([O])cc7)C([N][C@@H](CC([O])=O)C([N][C@@H](CCC([O])=O)C([N][C@H]1[C@@H](C)[O])=O)=O)=O)=O)=O)=O)=O)C(C)C)=O)=O)=O)=O)=O)=O)=O)=O)=O"
mol=Chem.MolFromSmiles(smi)
frags = break_cyclic_peptide_advanced(mol)
frag_smis = [ Chem.MolToSmiles(frag) for frag in frags ]
print(frag_smis)
输出:
['[1*][N][C@H](C([1*])=O)[C@@H](C)CC', '[1*][N][C@@H](CCCC[N])C([1*])=O', '[1*][N][C@@H](CCC[N]C([N])=[N])C([1*])=O', '[1*][N]C(Cc1c[nH]cn1)C([1*])=O', '[1*][N][C@H](C([1*])=O)[C@@H](C)[O]', '[1*][N][C@@H](Cc1ccccc1)C([1*])=O', '[1*][N][C@@H](CC(C)C)C([1*])=O', '[1*][N][C@@H](Cc1c[nH]c2ccccc12)C([1*])=O', '[1*][N][C@@H](C)C([1*])=O', '[1*][N][C@@H](CCSC)C([1*])=O', '[1*]C(=O)[C@@H]1CCCN1[1*]', '[1*][N][C@@H](C[S])C([1*])=O', '[1*][N][C@@H](CC([N])=O)C([1*])=O', '[1*][N][C@H](C([1*])=O)C(C)C', '[1*][N]CC([1*])=O', '[1*][N][C@@H](C[O])C([1*])=O', '[1*][N][C@@H](CCC([N])=O)C([1*])=O', '[1*][N][C@@H](Cc1ccc([O])cc1)C([1*])=O', '[1*][N][C@@H](CC([O])=O)C([1*])=O', '[1*][N][C@@H](CCC([O])=O)C([1*])=O'
可旋转二面角¶
提取分子中可旋转二面角,
示例文件: JGQ_ideal.sdf
示例代码:
import rdkit
from rdkit import Chem
from rdkit.Chem.Descriptors import NumValenceElectrons
from rdkit.Chem import Descriptors
from rdkit.Chem import AllChem
from rdkit.Chem.rdMolDescriptors import GetUSRScore, GetUSRCAT
import nglview
# https://files.rcsb.org/ligands/view/JGQ_ideal.sdf
sdffile="JGQ_ideal.sdf"
from rdkit import Chem
mols_suppl = Chem.SDMolSupplier(sdffile)
# print(type(mols_suppl))
mol=mols_suppl[0]
def getdihes(mol):
#pytmol label atom rank
dihes=[]
for bond in mol.GetBonds():
if bond.GetBondType().name=='SINGLE' and bond.IsInRing()==False:
atom2id=bond.GetBeginAtomIdx()
atom3id=bond.GetEndAtomIdx()
atom2=bond.GetBeginAtom()
atom3=bond.GetEndAtom()
if len(atom3.GetNeighbors())==1 or len(atom2.GetNeighbors())==1 :
pass
else:
atom1s=atom2.GetNeighbors()
atom1sid=[ at.GetIdx() for at in atom1s]
# print(atom1sid,atom3id)
atom1sid.remove(atom3id)
atom1id=atom1sid[0]
atom4s=atom3.GetNeighbors()
atom4sid=[ at.GetIdx() for at in atom4s]
# print(atom1sid,atom3id)
atom4sid.remove(atom2id)
atom4id=atom4sid[0]
# print(atom1id,atom2id,atom3id,atom4id)
dihe=[atom1id,atom2id,atom3id,atom4id]
dihes.append(dihe)
return dihes
dihes=getdihes(mol)
print(dihes)
输出:
[[18, 17, 29, 7], [6, 7, 29, 17], [8, 9, 10, 15], [21, 4, 26, 5], [14, 13, 16, 28], [26, 4, 21, 0], [21, 20, 25, 30]]
计算小分子的中心坐标¶
示例文件:
输入:
import rdkit
from rdkit import Chem
from rdkit.Chem.Descriptors import NumValenceElectrons
from rdkit.Chem import Descriptors
from rdkit.Chem import AllChem
from rdkit.Chem.rdMolDescriptors import GetUSRScore, GetUSRCAT
# import nglview
# 6IC_ideal.sdf
sdffile="5rec_lig.sdf"
sdffile="6IC_ideal.sdf"
from rdkit import Chem
mols_suppl = Chem.SDMolSupplier(sdffile)
mol3d=mol.GetConformer()
mol=mols_suppl[0]
# mol
def get_center(mol,mol3d):
coord=[0,0,0]
num=0
x=0
y=0
z=0
for ind,at in enumerate(mol.GetAtoms()):
num+=1
x+=mol3d.GetAtomPosition(ind).x
# print("x",x)
y+=mol3d.GetAtomPosition(ind).y
z+=mol3d.GetAtomPosition(ind).z
# print(x,y,z,num)
x=x/num
y=y/num
z=z/num
coord=[x,y,z]
return coord
get_center(mol,mol3d)
输出:
[0.11904545454545469, 0.03899999999999992, -0.004090909090909135]
计算描述符FSP3¶
Fsp3, the number of sp3 hybridized carbons/total carbon count。
from rdkit import Chem
from rdkit.Chem import AllChem, Descriptors, Descriptors3D, rdMolDescriptors
def mol_with_atom_index(mol):
for atom in mol.GetAtoms():
atom.SetAtomMapNum(atom.GetIdx())
return mol
smi="CCO[C@H]1CCN([C@@H](C1)c2ccc(cc2)C(=O)O)Cc3c4cc[nH]c4c(cc3OC)C"
mol=Chem.MolFromSmiles(smi)
mol_with_atom_index(mol)
FSP3=rdMolDescriptors.CalcFractionCSP3(mol)
print(FSP3)
输出
0.4
The RDKit Aromaticity Model¶
The Simple Aromaticity Model¶
The MDL Aromaticity Model¶
SMILES Support and Extensions¶
SMARTS Support and Extensions¶
Ring Finding and SSSR¶
Reaction SMARTS¶
Some features Chirality Rules and caveats
